从2019年12月8日开始,在我国武汉出现新型冠状病毒疾病(Corona Virus Disease 2019,COVID-19)。疫情迅速传播至全国,并在世界一定范围内传播。截至2月17日,我国新冠肺炎患者近6万多例。疫情的迅速传播使我国公共卫生和医疗资源受到了极大挑战,政府和社会各界高度重视和关注。该病毒基因组序列于2020年1月11日首次公布,被确认为一种新型冠状病毒(2019-nCoV),后被国际病毒分类学委员会(ICTV)命名为SARS冠状病毒2型(severe acute respiratory syndrome coronavirus 2,SARS-CoV-2)。现已明确其具有强烈的人传人能力,传播能力目前认为和SARS相当[1-4]。新冠状病毒传播方式主要包括飞沫传播和接触传播,潜伏期1-14天,多为3-7天[5]。发病症状有发热、咳嗽、肌痛、疲劳、呼吸困难,偶有咳痰、头痛、咯血等[6]。自其爆发以来,对病原溯源的研究一直受到社会各界广泛关注,但目前尚无定论。为了深入了解该病毒并从源头上阻断疫情传播,恢复正常生产生活,我们查阅了疫情爆发以来国内外SARS-CoV-2有关的科研论文,就其起源做一总结。
生物学特征
1.1形态及分类地位
冠状病毒(Coronaviruses,CoVs),属于巢病毒目(Nidovirales)冠状病毒科(Coronaviridae),在自然界中广泛存在,病毒颗粒呈球形或椭圆形,具有多形性,直径约60~220nm。病毒有包膜(envelope),包膜上存在棘突(spike),内部基因组为单股正链RNA (+ssRNA),病毒整个病毒颗粒在电镜下像皇冠或日冕并由此得名(图1)。

图1. 冠状病毒颗粒模式图和电镜图
冠状病毒科分为α,β,γ和δ四个属,可感染哺乳类人、鼠、猪、猫、犬、蝙蝠、牛和禽类脊椎动物,引起宿主呼吸道、肠道、肝和神经系统疾病,其中又以β-CoVs 对人类危害最严重[7-8]。β冠状病毒属分5个亚属,即Embecovirus、Sarbecovirus、Merbecovirus、Nobecovirus 和Hibecovirus[9]。人冠状病毒(HCoVs)于1965年首次被报道,它是所有年龄段普通感冒的病原之一,冬季和春季感染的发生率高[8]。引发此次疫情的新型冠状病毒于2020年1月6日被分离出,电镜下该病毒形态和冠状病毒的形态一致。基于宏基因组学测序拼接得到其全基因组序列,系统进化分析发现分类学为冠状病毒β属Sarbecovirus亚属,属于从未被发现过的新型冠状病毒,其进化距离明显远于2002年11月我国爆发的SARS (severe acute respiratory syndrome, SARS)和2012年中东及韩国等地爆发的MERS (Middle East Respiratory Syndrome)病原体[10-12]。SARS-CoV-2是目前已知的第7种可以感染人的冠状病毒,其余6种分别是HCoV-229E、HCoV-OC43、HCoV-NL63、HCoV-HKU1、SARS-CoV和MERS-CoV [7-8]。
1.2基因组结构
冠状病毒遗传物质是所有RNA病毒中最大的,基因组全长在26~32 kb 之间。病毒基因组5'端约2/3为重叠的开放阅读框(Open reading frame,ORF) ORF1a 和ORF1b,主要负责编码与病毒复制和转录相关的酶等非结构蛋白。后面的1/3 基因组负责编码刺突蛋白(Spike protein,S)、小膜蛋白(Envelope membrane protein,E)、膜蛋白 (Membrane protein,M)和核衣壳蛋白(Nucleocapsid protein,N)等主要结构蛋白,另外还有嵌套在3'端基因组中的一系列基因编码辅助性蛋白[8-9]。这其中结构蛋白至关重要,决定了病毒的组装、稳定性和侵袭力。
现已测定了来自不同省份COVID-19患者的几十株SARS-CoV-2全基因组序列,序列间高度保守,少量序列差异可能是早期在人群传播过程中适应性突变产生的。有研究[14]通过对首次公布的SARS-CoV-2基因组进行了注释,发现其编码至少27种蛋白质,其中包括15种非结构蛋白(nsp1-nsp10,nsp12-nsp16),4种结构蛋白(S,E,M和N)以及8种附属蛋白(3a, 3b,p6,7a,7b,8b,9b和14),基因组类似SARS-CoV(图2A),但明显存在一些编码蛋白质序列的插入和缺失。尤其是S蛋白,SARS-CoV-2编码的S蛋白序列组成和SARS以及MERS相比差异最大。SARS-CoV-2和SARS-CoV的S蛋白均可分为S1和S2结构域[15]。在病毒入侵宿主细胞过程中,宿主源组织蛋白酶切割S蛋白后得到一个S1受体结构域,内含受体结合区(receptor binding domain,RBD)可与来自不同动物的血管紧张素转换酶Ⅱ (angiotensin converting enzyme Ⅱ,ACE2,在人体中主要分布于呼吸道上皮细胞、肺脏、心脏、肾脏和消化道等位置)作为受体来感染宿主[16-17],此过程激活了病毒感染力并决定了宿主范围[18-19];另外得到一个主要介导病毒包膜与细胞膜融合的S2结构域。SARS-CoV-2和SARS-CoV在S蛋白的RBD区与宿主受体之间结合的5个关键位点中,有4个是不同的,这些不同的位点是新型冠状病毒在致病能力上与SARS冠状病毒有差异的潜在原因(图2B)。
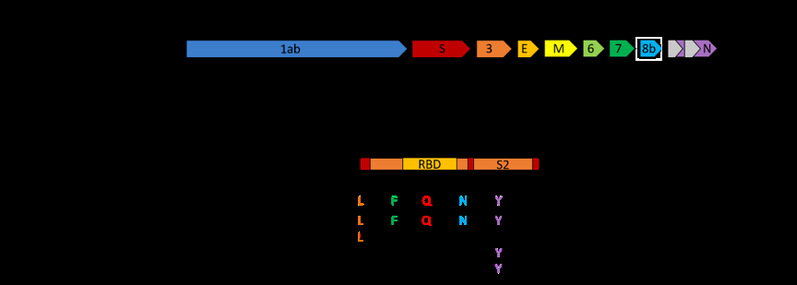
图2. (A) SARS-CoV-2冠状病毒基因组结构及其与 (B) 近源冠状病毒在S蛋白RBD序列部分关键位点的差异
2. SARS-CoV-2宿主与进化
2.1自然宿主
SARS-CoV-2起源研究可以参考SARS相关内容。研究证明[20]SARS冠状病毒是通过蝙蝠-果子狸-人实现传播的,尤其是2013年,SARS冠状病毒被溯源到中华菊头蝠(Rhinolophus sinicus)[7]。武汉华南海鲜市场相关人员出现聚集性感染后,最初对SARS-CoV-2的溯源曾认为该地就是病毒爆发起源地。然而有证据表明第一批41个确诊COVID-19病例中总共有13个病例与华南海鲜市场无接触史;在更早出现的4名感染者中,有3人没有华南海鲜市场暴露史,因此海鲜市场不是唯一暴露源[6]。结合新冠状病毒相对较长的潜伏期,可以推断最早的感染可能发生在2019年11月。SARS-CoV-2首次从一个在武汉华南海鲜市场工作的感染者支气管肺泡灌洗液中利用宏基因组测序拼接出来,系统发育分析首次发现该病毒和蝙蝠来源的SARS样冠状病毒(SARS-like CoV)紧密聚在β冠状病毒属,Sarbecovirus亚属[21],由此开启病毒溯源系列研究。
在这之后,有研究者将公布的新型冠状病毒序列与不同来源的冠状病毒序列进行了系统发育分析,确认了新型冠状病毒SARS-CoV-2与SARS和SARS类冠状病毒的亲缘关系,推测自然宿主可能是蝙蝠。对SARS-CoV-2 的S蛋白进行的结构建模揭示了其RBD结合区与人类ACE2分子受体有极强的相互作用,提示病毒可能通过S-ACE2结合途径传播并构成重大的公共卫生风险[22]。随后,有研究报道蛇来源的病毒和新病毒的同义密码子使用偏好性(Relative Synonymous Codon Usage, RSCU)类似,并以此推测蛇是病毒储存宿主[23]。然而尚无冠状病毒可以感染蛇的证据,基于此方法的重分析也没有支持该推测[24]。
紧接着,有研究[25]通过高通量测序获得了多条全基因组序列,发现相比于与SARS-CoV(约79%)、MERS-CoV(约50%)的同源性,SARS-CoV-2全基因组与浙江舟山的两株蝙蝠样冠状病毒株Bat-SL-CoVZC45(同源性87.99%)和Bat-SL-CoVZXC21(同源性87.23%)进化关系更近,但SARS-CoV-2的进化分支长度更长,说明新病毒进化上更晚出现。同源建模同样也显示,尽管关键氨基酸残基存在差异(图2B),SARS-CoV-2和SARS-CoV具有相似的受体ACE2结合区。该研究同时推测中间宿主可能在华南海鲜市场出售的动物中[25]。
最终,一项研究将SARS-CoV-2基因组序列与该实验室早期从云南采集到的中菊头蝠(Rhinolophus affinis)体内检测到的Bat-Cov-RaTG13冠状病毒株基因组序列进行比较,发现两者全基因组水平一致性高达96.2%,同时也发现两者的S基因片段差异性最大(基因同源性为93.1%)[27]。这一研究直接将SARS-CoV-2的自然宿主溯源工作推进到中菊头蝠,该结论得到了同行研究的支持[26]。另外,以上研究还通过实验验证了新型冠状病毒能感染表达除鼠以外的人、猪、中华马蹄蝠(Chinese horseshoe bats)/中华菊头蝠(Rhinolophus sinicus)和果子狸(civet)等物种的ACE2受体的非敏感细胞[27]。
除此以外,其它一些相关研究也都支持SARS-CoV-2的自然宿主就是中菊头蝠。比如有研究者使用β冠状病毒基因组中的一个互补回文序列对新型冠状病毒基因组进行溯源,结果也和中菊头蝠匹配[28]。另有发现多条已公布序列中含有不同于其他SARS类冠状病毒的“RRAR”序列,可供Furin蛋白酶切(识别并切割“RXXR”模式增加膜融合能力)的位点,并推测这个位点有可能增强了SARS-CoV-2的感染和传播能力。当然,“RRAR”序列在新冠状病毒中的功能尚需反向遗传学实验验证[29]。
2.2中间宿主
目前的证据虽然都支持SARS-CoV-2的自然宿主是中菊头蝠,然而Bat-CoV-RaTG13和SARS-CoV-2还是有1100多个碱基序列是不一致的。这种差异性不太容易导致病毒从Bat-CoV RaTG13直接感染给人,需要通过在进化地位处于蝙蝠和人中间的某一个物种(也可能是多种)作为中间宿主,在中间宿主中进化后才能更适于感染人。比如果子狸来源的冠状病毒与人类SARS病毒之间的差异仅为29个核苷酸序列[30],因此SARS的源头宿主是蝙蝠,但要通过中间宿主果子狸才能传染人;MERS的源头宿主也是蝙蝠,中间宿主是骆驼(图3)。另外,冬季蝙蝠在山洞中冬眠,很难直接感染人。因此,中间宿主才是病毒的直接传染源。然而,SARS-CoV-2中间宿主的寻找过程似乎更扑朔迷离。
在SARS-CoV-2的溯源中,有一研究比较了脊椎动物宿主的病毒传染模式,发现水貂(mink)体内冠状病毒的传染模式更接近新型冠状病毒[31]。该研究提示水貂有作为中间宿主的可能性。然而,最新的一项研究通过广泛的宏基因组数据库搜索发现,与新型冠状病毒序列匹配上最多的是穿山甲肺组织来源的冠状病毒测序片段[32]。无独有偶,另外有一研究从马来亚穿山甲(Manis javanica)冠状病毒宏基因组测序数据库中重建出一条全基因组序列,后经过比对发现和新型冠状病毒基因组序列相似性为90.5%。令人惊讶的是,该穿山甲冠状病毒与SARS-CoV-2在S蛋白的RBD区的进化关系在已知病毒中最为保守(基因同源性89%,氨基酸同源性为98%),高于RaTG13与新病毒RBD区的同源性(基因同源性75%,氨基酸同源性78%),同时两者在RBD和ACE2受体结合的5个关键氨基酸位点和新型冠状病毒完全一致(图2B)[33]。由此可见SARS-CoV-2的RBD序列很可能就是从马来亚穿山甲体内冠状病毒获得的,进而可以推测穿山甲是目前看来最可能的中间宿主。
其实早在COVID-19疫情爆发前的2019年10月,已有报道从广东海关缉私的马来亚穿山甲死亡个体的甲肺、淋巴和脾脏等器官通过病毒宏基因组测序得到的多个片段重叠群,比对数据库得到多种SARS相关冠状病毒[34]。这佐证了马来亚穿山甲作为SARS-CoV-2中间宿主的可能性。由SARS-CoV-2中间宿主的追踪过程可见野生动物病毒数据库完整度的重要性,同时也说明用病毒基因组中功能片段分别去匹配数据库的重要性,这些都是能找到潜在中间宿主的关键。
2.3 SARS-CoV-2是重组病毒吗?
冠状病毒基因组较大,作为RNA单链不稳定,且复制过程中没有校正机制,导致冠状病毒变异快、宿主多且具有较强的宿主适应性等特点(更好的结合细胞受体、复制和毒力增强等)[35]。SARS-CoV基因组上不同序列在进化上来源不同,被认为是先在几个蝙蝠SARS样冠状病毒之间进行基因重组[36],之后又嵌入了其它哺乳动物来源的病毒基因序列-嵌合进化论(Mosaic Evolution) [37]。
穿山甲虽然是国家二级保护动物,但是民间作为中药材已有相当长的历史,难以解释为什么之前没有引起人类感染。穿山甲喜欢钻山洞,容易接触到山洞里具有感染性的蝙蝠粪便[38]。那么可以设想:SARS-CoV-2的产生有可能是马来亚穿山甲感染了中国西南菊头蝠携带的冠状病毒,菊头蝠来源的冠状病毒基因组与马来亚穿山甲体内的冠状病毒通过重组交换获得S基因的RBD序列从而产生了重组体。该重组病毒在穿山甲体内进行一定时间正向选择的适应性进化获得了更好的稳定性和侵染能力,从而演变成了SARS-CoV-2。马来亚穿山甲冠状病毒基因序列相比氨基酸序列极其不保守[33],说明有高水平核苷酸替换率,在选择压力下病毒快速地变异和进化。这也许可以解释为什么SARS-CoV-2引起的疾病临床症状与2003年爆发的SARS显著不同,死亡率约为SARS的1/3,但潜伏期更长[39]。关于SARS-CoV-2出现的时间,基于GTR+G+I的随机替换模型,有研究通过贝叶斯联合系统发育分析多条新病毒全基因组序列,计算核苷酸替换率并得出SARS-CoV-2 进化上最近的共同祖先在2019年12月8日爆发前91-214天就已经出现了[40]。
回顾性研究发现,2002 年首例SARS患者是广东省深圳一家餐厅的厨师,其供职的餐馆有包括蝙蝠在内的各种野味[41]。由此可以假设SARS-CoV-2首例感染者可能和马来穿山甲往武汉华南海鲜市场运输贩卖有关,当然这需要提供更多的全基因组序列信息并通过建立合适的进化分析模型进行深入研究。另外,疫情爆发地武汉市江河湖泊众多,2019年末气候相对温暖和湿润,这种气候是否促进了重组病毒的产生和传播也需要进一步研究。
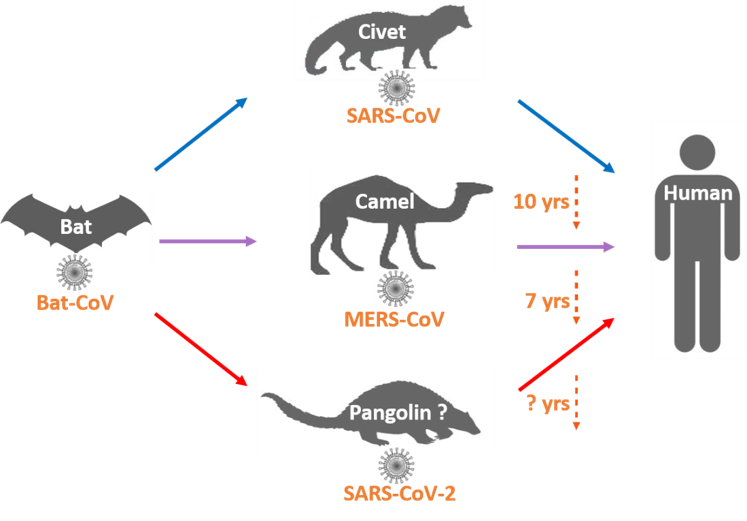
图3. SARS-CoV,MERS-CoV和SARS-CoV-2传播示意图
3结语
查找传染源和病原体溯源是应对重大公共卫生事件的首要任务和关键环节。虽然研究发现穿山甲是潜在的SARS-CoV-2的中介, 但也不能排除有其他中间宿主的可能。在正常的生态环境下,人类和蝙蝠、穿山甲等野生动物一直和平相处,野生动物的贩卖交易导致新型病毒直接与人类的接触并构成了威胁。当前,我国城市化进程持续加速,人员流动日益频繁,各种RNA病毒的累积突变和重组变异产生新型病毒并造成人间大流行将随时可能发生。因此严格管理野生动物市场,加强动物检疫,防止新型病毒的重新爆发。关于SARS-CoV-2未来应获得更多冠状病毒的基因变异样本,计算病毒进化速率,应密切监测病毒是继续向强毒性还是弱毒性演变。另外,还应该通过细胞和动物水平上的实验重点研究S蛋白RBD结构域的突变与ACE2蛋白适合性或致病性的关系,掌握该病毒的变异和致病机制,提前研发相关疫苗和抗病毒药物作为储备以防新一轮的冠状病毒爆发。除此以外,还应该建立健全野生动物携带的病毒基因组数据库,便于新发传染病病原体的预警和及时溯源,同时加强实验室安全管理,谨防类似SARS实验室泄露事件的发生。
参考文献
[1] Chan JF, Yuan S, Kok KH, et al .A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster[J].Lancet, 2020, 395(10223):514-523. DOI: 10.1016/S0140-6736(20)30154-9.
[2] Li Q, Guan X, Wu P, et al .Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus –Infected Pneumonia[J].N Eng J Med,2020, DOI:10.1056/NEJMoa2001316.
[3] Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 Hospitalized Patients with 2019 novel coronavirus-infected Pneumonia in Wuhan, China[J].JAMA,2020, DOI:10.1001/jama.2020.1585.
[4] Riou J ,Althaus CL. Pattern of early human-to-human transmission of Wuhan 2019 novel coronavirus (2019-nCoV), December 2019 to January 2020[J]. Euro Surveill, 2020, 25(4), DOI:10.2807/1560-7917.ES.2020.25.4.2000058.
[5] 国家卫生健康委办公厅.新型冠状病毒感染的肺炎诊疗方案(试行第五版)[Z]. 2020年2月4日印发.
General office of the national health commission. Pneumonia diagnosis and treatment plan for new coronavirus infection (trial version 5) [Z]. Issued on February 4, 2020.
[6] Chaolin Huang, Yeming Wang, Xingwang Li, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China[J/OL]. Published Online January 24, 2020 https://doi.org/10.1016/ S0140-6736(20)30183-5.
[7] CORMAN VM, MUTH D, NIEMEYER D, et al. Hosts and Sources of Endemic Human Coronaviruses [J]. Adv Virus Res, 2018, 100:163-188, DOI: 10.1016/bs.aivir.2018.01.001.
[8] CUI J, LI F, SHI ZL. Origin and evolution of pathogenic coronaviruses [J]. Nat Rev Microbiol, 2019, 17(3):181-192, DOI: 10.1038/s41579-018-0118-9.
[9] International Committee on Taxonomy of Viruses, Taxonomy History: Cornidovirineae [EB/OL].https://talk.ictvonline.org/taxonomy/p/taxonomy-history?taxnode_id=20186105 (accessed on 2 January 2020).
[10] KOH D, SNG J. Lessons from the past: perspectives on severe acute respiratory syndrome [J]. Asia Pac J Public Health, 2010, 22(3 Suppl): 132S-136S, DOI: 10.1177/1010539510373010.
[11] WHO. Current outbreak situation in the Republic of Korea and China as of 17 August 2015[EB/OL]. (2015-8-17).
[12] CHOI JY. An outbreak of Middle East respiratory syndrome coronavirus infection in South Korea, 2015 [J]. Yonsei Med J, 2015, 56: 1174-1176, DOI: 10.3349/ymj.2015.56.5.1174.
[13] Song, Z., Xu, Y., Bao, L., et al. (2019). From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses [J/OL]. 11(1). Published online 2019/01/17 DOI: 10.3390/v11010059.
[14] Wu A, Peng Y, Huang B, et al. Genome composition and divergence of the novel coronavirus(2019-nCoV)originating in China[J].Cell Host Microbe, 2020, DOI:10.1016/j.chom.2020.02.001.
[20] Chinese SARS Molecular Epidemiology Consortium. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. [J].Science, 2004,303 (5664): 1666-1669, DOI: 10.1126/science.1092002.
[21] Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China [J]. Nature, 2020, DOI: 10.1038/s41586-020-2008-3.
[22] Xu X, Chen P, Wang J, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission [J]. Life Sciences, 2020, doi: 10.1007/s11427-020-1637-5.
[23] Ji W, Wang W, Zhao X, et al. Homologous recombination within the spike glycoprotein of the newly identified coronavirus may boost cross-species transmission from snake to human [J]. MEDICAL WIROLOGY, 2020, doi:10.1002/jmv.25682.
[24] Chengxin Zhang, Wei Zheng, Xiaoqiang Huang, et al. Protein structure and sequence re-analysis of 2019-nCoV genome does not indicate snakes as its intermediate host or the unique similarity between its spike protein insertions and HIV-1[J]. q -bio, 2020, Cite as: arXiv: 2002. 03173 [q-bio.GN].
[25] Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding [J]. Lancet, 2020, doi: 10. 1016/ S0140-6736(20)30251-8.
[26] Paraskevis D, Kostaki EG, Magiorkinis G, et al.Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event [J]. Infect Genet Evol, 2020, 79: 104212, DOI: 10.1016/j.meegid.2020.104212.
[27] Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin [J].Nature, 2020, DOI: 10.1038/s41586-020-2012-7.
[28] 陈嘉源, 施劲松, 丘栋安等. 武汉2019 冠状病毒基因组的生物信息学分析[J].生物信息学, 1672-5565(2020)02-000-00.
Chen Jia-yuan, Shi Jin-song, Qiu Dong-an et al. Bioinformatics analysis of coronavirus genome in wuhan 2019 [J]. Bioinformatics, 1672-5565 (2020) 02-000-00.
[29] 李鑫, 段广有,张伟等. 武汉2019 冠状病毒S 蛋白可能存在Furin 蛋白酶切位点.生物信息学[J], 1672-5565(201x)03-000-00.
Li Xin, Duan Guangyou, Zhang Wei et al. Possible Furin proteinase cleavage site for S protein of wuhan 2019 coronavirus. Bioinformatics [J], 1672-5565 (201x) 03-000-00.
[30] Guan Y, Zheng BJ, He YQ, et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China [J]. Science, 2003. 302(5643): 276-278, DOI: 10.1126/science.1087139.
[31] Qian Guo, Mo Li, Chunhui Wang, et al. Host and infectivity prediction of Wuhan 2019 novel coronavirus using deep learning algorithm [J]. bioRxiv, 2020, doi: https://doi.org/10.1101/2020.01.21.914044.
[32] Lamia Wahba, Nimit Jain, Andrew Z Fire, et al. Identification of a pangolin niche for a 2019-nCoV-like coronavirus through an extensive meta-metagenomic search [J]. bioRxiv, 2020, https://doi.org/10.1101/2020.02.08.939660.
[33] Matthew C. Wong, Sara J. JavornikCregeen, et al. Evidence of recombination in coronaviruses implicating pangolin origins of nCoV-2019[J]. bioRxiv, 2020, https://doi.org/10.1101/2020.02.07.939207.
[34] Liu P, Chen W, Chen JP. Viral Metagenomics Revealed Sendai Virus and Coronavirus Infection of Malayan Pangolins (Manis javanica) [J].Viruses, 2019, 11(11):979, DOI: 10.3390/v11110979.
[35] Cui J, Li F, Shi ZL.Origin and evolution of pathogenic coronaviruses [J]. Nat Rev187 Microbiol, 2019, 17:181-192, DOI: 10.1038/s41579-018-0118-9.
[36] Hu B, Zeng LP, Yang XL, et al. Discovery of a rich gene pool of bat SARS related coronaviruses provides new insights into the origin of SARS coronavirus[J] PLoS Pathog, 2017, 13(11), https://doi.org/10.1371/journal.ppat.1006698.
[37] John, Stavrinides, David S, Guttman. Mosaic Evolution of the Severe Acute Respiratory Syndrome Coronavirus [J]. Journal of virology, 2020, 78 (1):76-82, DOI: 10.1128/jvi.78.1.76-82.2004.
[38] WATANABE S, MASANGKAY JS, NAGATA N, et al. Bat coronaviruses and experimental infection of bats, the Philippines [J]. Emerg Infect Dis, 2010, 16, 1217-1223, DOI: 10.3201/eid1608.100208.
[39] Yang Yang, Qingbin Lu, Mingjin Liu, et al. Epidemiological and clinical features of the 2019 novel coronavirus outbreak in China.Medrxiv, Posted February 11, 2020, doi.org/10.1101/2020.02.10.20021675.
[40] Chenglong Xiong, Lufang Jiang, Yue Chen, et al.Evolution and variation of 2019-novel coronavirus [J]. doi: https://doi.org/10.1101/2020.01.30.926477.
[41] ZHAO GP. SARS molecular epidemiology: a Chinese fairy tale of controlling an emerging zoonotic diseasein the genomics era [J]. Philos Trans R Soc Lond B Biol Sci, 2007, 362(1482): 1063-1081,DOI: 10.1098/rstb.2007.2034.